A 31-year-old male applies for life insurance. He states that he is in good health except for hypertension (current and treated) and a diagnosis of Henoch-Shőnlein Purpura at the age of 27 years. His medical records reveal that he presented with a rash, hematuria, trace proteinuria and diarrhea. The rash and diarrhea resolved in about one month, but the hematuria persisted for about 12 months.
Urology has been following him every six months, and for the last three visits, he has had normal blood pressure (treated), no proteinuria and no hematuria neither macroscopic nor microscopic, along with normal renal function studies. His insurance labs have the same normal results including liver function tests, renal function tests and urinalysis.
What are the mortality considerations for someone with a diagnosis of Henoch-Shőnlein Purpura?
Definition
Henoch Shőnlein Purpura (HSP) is an acute small vessel vasculitis and the most common vasculitis in children. 90% of cases occur in children younger than 10 years old. Incidence is between 10-20 per 100,000 children less than 17 years old.
Adult HSP, in contrast, is less common but often is associated with a worse clinical course and outcome. In some areas of the world, as many as 25%-30% of patients with HSP are adults.
The clinical features of HSP are atypical at the extremes of age, with adults being more severely affected and children under 2 years old being mildly affected. HSP occurs primarily in the fall to spring months and often follows an upper respiratory infection,
although other pathogens, drugs and environmental factors have been implicated as triggers.
HSP is often, though not always, self-limited. It may eventually develop into chronic kidney disease with the long-term prognosis depending on the severity of the renal
involvement.
HSP was renamed IgA Vasculitis (IgAV) by the International Chapel Hill Consensus Conference Nomenclature of Vasculitides in 2012, yet the new name is not universally used, and in this article, will be referred to as HSP.
Presentation
The presentation varies, but universally, a non-thrombocytopenic palpable purpura mostly located on the buttocks and lower extremities is present either initially or shortly into the course. Any of the following may also be present: arthritis, abdominal pain and renal involvement.
Diagnostic criteria
Although there is more than one set of criteria in existence, the most universally used consensus criteria were developed by the European League Against Rheumatism (EULAR) and the Paediatric Rheumatology European Society (PRES) and validated
in conjunction with the Paediatric Rheumatology International Trials Organisation (PRINTO).
| Mandatory Criteria: | Must have one or more of the following: |
Rash (Purpura), usually
palpable and in clusters with lower limb predominance and without thrombocytopenia or coagulopathy | Abdominal pain (usually diffuse with acute onset) |
| Arthritis or arthralgia |
| Renal involvement (proteinura, hematuria) |
| Leukoclastic vasculitis or proliferative glomerulonephritis with predominant IgA deposition |
There are no current lab values that are diagnostic for HSP. To receive this diagnosis, platelet levels must be normal or elevated, they cannot be low. Common findings might include: leukocytosis, elevated Erythrocyte Sedimentation Rate (ESR), elevated IgA
serum levels, hematuria, positive throat culture for beta-hemolytic strep or low complement levels.
In cases where the diagnosis cannot be made reliably on clinical findings and routine laboratory values, the presence of leukocytoclastic vasculitis and IgA-immune deposits on skin or on kidney biopsies point strongly toward HSP as the diagnosis.
There is some exciting research on the horizon with the possible identification of a urinary protein as a marker for HSP nephritis. A definitive marker would be especially beneficial for those presentations which are atypical or cases in which a biopsy would be difficult.
Clinical course and differential diagnosis
Cutaneous palpable purpura are an essential feature in this illness (see Figure 1). They are most prominent in the buttocks and lower extremities but may present elsewhere. There may also be scattered petechiae and coalescing ecchymosis.
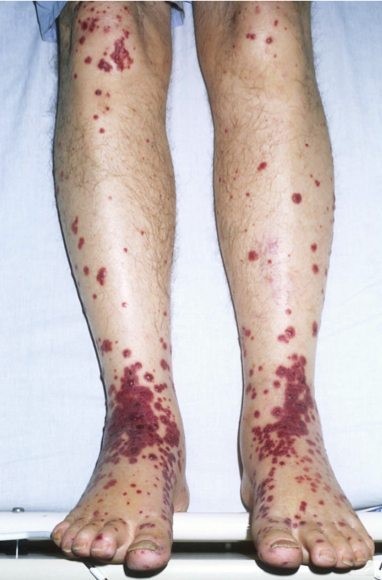
This is differentiated from immune thrombocytopenia purpura (ITP) simply by checking a platelet count. The platelet count will be low in ITP. Other vasculitides would need to be considered, especially if the illness presents in a piecemeal fashion.
Arthritis and arthralgias are the second leading feature in this condition. These most often involve the joints of the lower extremities, making it painful to walk. In some cases, young children simply refuse to walk. Arthritis will precede the skin findings by up to one week in 15%-25% of cases.
Gastrointestinal involvement occurs in 50%-75% of patients. Colicky abdominal pain, diarrhea, vomiting and gastrointestinal bleeding are the predominant symptoms. Guaiac positive stools are found in >50% of patients with HSP.
If these symptoms predate the rash (they do in 10%-20% of patients), as they can by one to two weeks, other causes of abdominal pain must be ruled out. GI bleeding is not uncommon and about 30% of HSP patients will have melanotic stools.
Renal involvement occurs in 40%-50% of patients with microscopic hematuria being most common followed by proteinuria and gross hematuria. Proteinuria accompanies hematuria 60% of the time but is rarely the sole renal finding.
Nephritis rarely if ever precedes the purpura, predominantly manifests within four weeks of the skin finding and nearly always within three months of the presentation of the other symptoms. As a result, urinalysis should be done weekly during the active disease and then each month for three months. If at any time there is evidence of nephritis, long term monitoring of urine, renal function and blood pressure is warranted.
Differences between adult and children with HSP
A 10-year retrospective study was done in Korea to investigate the differences in clinical manifestations and outcomes between adult and child patients with HSP.
| FINDINGS | ADULTS | CHILDREN |
| Purpura in upper extremities | 41.7% | 19.3% |
| Arthralgias | 27.1% | 55.4% |
| Anemia | 25.0% | 7.1% |
| Diarrhea | 20.0% | 1.6% |
| Elevated serum IgA | 26.3% | 3.5% |
| Renal involvement | 79.2% | 30.4% |
| Chronic renal failure at follow up | 10.4% | 1.8% |
These findings are in line with previous comparisons between children and adults with HSP. Generally, adults tend to have slightly different non-skin symptoms as well as more serious renal manifestations, both acutely and chronically.
Treatment
Treatment is generally supportive as the disease course is usually self-limited. The rare cases of bowel perforation or intussusception would require surgical intervention. Corticosteroids early in the course tend to alleviate gastrointestinal and joint symptoms but do not prevent delayed nephritis.
There are a number of different treatments used for HSP with moderate to severe renal involvement, including steroids and adjuvant treatments such as cyclophosphamide, enalapril, dipyridamole and IVIG. There is no clearly superior treatment, and
close monitoring is required.
Prognosis
Symptoms last, on average, four weeks. Recurrences are not unusual and generally subside after six months. Renal signs and symptoms such as hematuria and proteinuria can persist for months to years
Fortunately only 1%-3% of children and about 10% of adult patients progress to end stage kidney disease. Microscopic hematuria with trivial proteinuria portends a favorable prognosis, while nephritis complicated by nephrotic syndrome as well as >50% crescent
formation on renal biopsy suggest a less favorable prognosis.
Current research
While the pathogenesis of HSP is still largely unknown, the disease is characterized by IgA1 immune deposits, complement factors and neutrophil infiltration accompanied by vascular inflammation.
HSP nephritis—also known as IgA vasculitis with nephritis (IgAVN)—resembles IgA nephropathy (IgAN). Both are associated with galactose-deficient IgA1 deposits in the kidney. Several labs across the world are currently working to better delineate the
pathogenesis and aid in the treatment.
Returning to the case
As this gentleman was an adult when he developed HSP, his renal status needs to be assessed and monitored. While he had hematuria and trace proteinuria at presentation, he has been well followed by urology and appears to no longer have the renal manifestations of HSP. His labs and clinical course seem to put him in the fortunate category of no long term renal sequelae and, therefore, no expected excess mortality.
References
Calviño, Maria C., et al. “Henoch-Schönlein Purpura in Children from Northwestern Spain: a 20-year Epidemiologic and Clinical Study.” Medicine 80.5 (2001): 279-290.
Chan, Han, et al. “Risk Factors Associated with Renal Involvement in Childhood Henoch-Schönlein Purpura: a Meta-analysis.” PloS One 11.11 (2016): e0167346.
Heineke, Marieke H., et al. “New Insights in the Pathogenesis of Immunoglobulin-A Vasculitis (Henoch-Schönlein Purpura).” Autoimmunity Reviews (2017).
Jennette, John C., et al. “2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides.” Arthritis & Rheumatology 65.1 (2013): 1-11.
Kang, Yoon, et al. “Differences in Clinical Manifestations and Outcomes Between Adult and Child Patients with Henoch-Schönlein Purpura.” Journal of Korean Medical Science 29.2 (2014): 198-203.
López-Mejías, Raquel, et al. “Genetics of Immunoglobulin-A Vasculitis (Henoch-Schönlein Purpura): An Updated Review.” Autoimmunity Reviews (2018).
Pillebout, Evangéline, et al. “Henoch-Schönlein Purpura in Adults: Outcome and Prognostic Factors.” Journal of the American Society of Nephrology 13.5 (2002): 1271-1278
Saulsbury, Frank T. “Clinical Update: Henoch-Schönlein Purpura.” The Lancet 369.9566 (2007): 976-978.
Suzuki, Hitoshi, et al. “IgA Nephropathy & IgA Vasculitis with Nephritis Have a Shared Feature Involving Galactose-Deficient IgA1-oriented Pathogenesis.” Kidney international
(2018).
Wang, Jiapei, et al. “Elevated Urinary Monocyte Chemoattractant Protein-1 Levels in Children with Henoch-Schonlein Purpura Nephritis.” Pediatrics & Neonatology(2017).
Yang, Yao-Hsu, Hsin-Hui Yu, and Bor-Luen Chiang. “The Diagnosis and Classification of Henoch–Schönlein Purpura: An Updated Review.” Autoimmunity Reviews 13.4-5 (2014):
355-358.
Zaffanello, Marco, et al. “Adjuvant Treatments for Henoch-Schönlein Purpura Nephritis in Children: A Systematic Review.” Current Therapeutic Research 70.3 (2009): 254-265.
Up To Date, last accessed 2/14/2018